












研究背景
坏死性凋亡是一种程序性形式的坏死或炎性细胞死亡,它不仅有助于病原体的清除,而且还能导致疾病的发病。坏死性凋亡是由RIPK 3介导的MLKL磷酸化触发的,这被认为会引发MLKL寡聚化、膜移位和膜破裂,但其确切机制尚不完全清楚。
研究结果
1.MLKL在坏死性凋亡期间泛素化
通过监测MLKL在坏死触发因子作用下的泛素化状态,研究了MLKL介导的坏死性凋亡的调节机制。结果显示,虽然用TNF/SMAC模拟物(SM)/z-VAD-FMK (TSZ)治疗在人结直肠癌HT-29细胞、小鼠皮肤成纤维细胞(MDFs)和小鼠L929细胞中引起时间和RIPK1依赖性坏死,但TSZ也在所有这些细胞类型中触发内源性MLKL的泛素化(图1a-d);用非特异性去泛素化酶USP21处理完全去除了MLKL的涂抹模式,证明MLKL被Ub加合物修饰以响应TSZ(图1c)。研究还发现MLKL泛素化程度与坏死程度相关,相应地,MLKL的磷酸化和泛素化有着相似的动力学(图1c,d),泛素化的MLKL被磷酸化(图1d,第二组),MLKL的泛素化不仅发生在TNF所致的坏死反应中,而且在TRAIL引起的死亡中也发生了变化(图1e,f)。这表明MLKL的泛素化是对各种坏死信号事件的反应。MLKL在TNF诱导的细胞凋亡过程中没有泛素化(图1g,h)。这些结果表明MLKL在对死亡刺激的反应中泛素化。

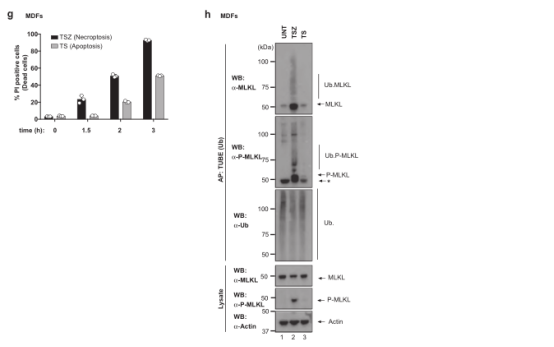
图1
2.MLKL的泛素化需要RIPK 3介导的磷酸化
通过评估MLKL泛素化是否需要RIPK1 > RIPK3信号传导,作者发现RIPK1和RIPK3的药理学抑制或RIPK3的遗传缺失不仅阻止了坏死性凋亡(图2a,b)还消除了MLKL的泛素化(图 2b,c),这些结果表明 MLKL 泛素化高度依赖于 RIPK3 的激酶活性。为了证明这一观点,测试了MLKL泛素化是否需要RIPK3介导的S345磷酸化,方法是用不能被RIPK3磷酸化的野生型(WT)或MLKL的突变形式(MLKL S345A)重建Mlkl -/- MDFs ,因此无法触发坏死性凋亡。
结果表明,无论S345处的MLKL磷酸化如何,都会出现微弱的、较高分子量的泛素化涂抹模式(图 2d),该结果与MLKL的活性磷酸化形式是泛素化的观点一致。此外,在评估MLKL的泛素化形式是否也包含在寡聚体中时发现MLKL低聚物也被泛素化。因此,与USP21孵育消除了Ub涂抹模式,导致出现MLKL的寡聚形式以及单体形式(图2f)。这些数据说明了磷酸化单体和寡聚体都已被泛素化,表明P-MLKL的泛素化是早期事件,并且发生在其寡聚化和膜易位之前。
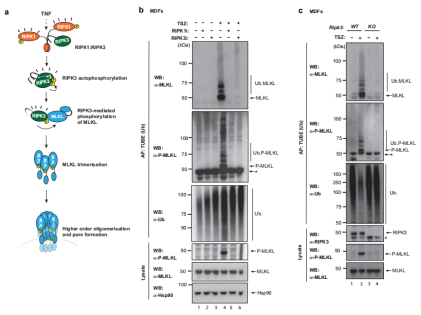
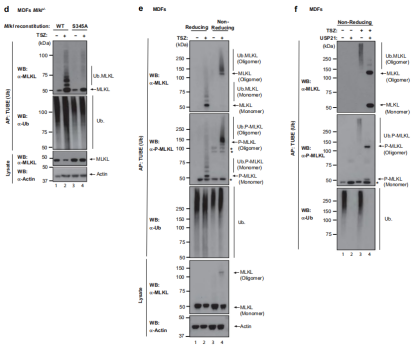
图2
3.MLKL在质膜定位之前被泛素化。
使用原位邻近连接测定(PLA)和亚细胞分级分离方法了解 MLKL的泛素化是否发生在质膜运输之前。使用针对MLKL和Ub的一抗,观察到了用TSZ处理引起显着的MLKL:Ub PLA斑点,而在未处理的对照条件下未检测到此类斑点(图 3a),通过进行细胞分级然后进行Ub富集,发现TSZ处理导致在2小时时在细胞溶质和膜富集部分中出现泛素化MLKL(图 3b)。这些结果支持MLKL在其易位到质膜之前主要在胞质区室中泛素化的观点。此外,使用多种Ub链选择性亲和试剂以确定与MLKL共轭的Ub链类型,发现坏死性凋亡刺激在MDFs和HT-29中MLKL触发了显著的k63连接的泛素化(图 3c,d),在这些条件下,没有检测到m1-或k48-连接的链。k63连接的Ub链的存在表明MLKL的泛素化具有信号传导功能并且不会触发其蛋白酶体降解。
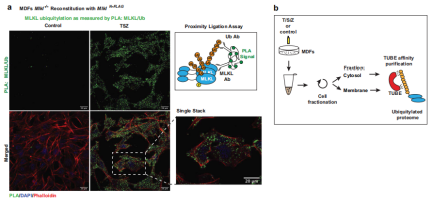
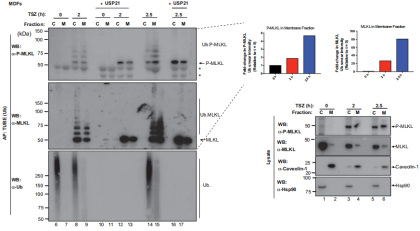
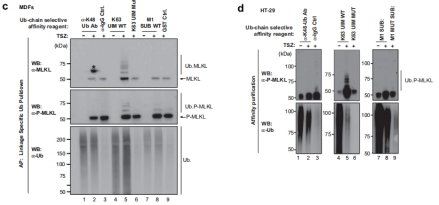
图3
4.内源性MLKL在K51、K77、K172和K219上泛素化
通过鉴定MLKL的Ub受体赖氨酸残基以研究MLKL泛素化在坏死性凋亡信号传导中的作用。研究结果确定了TNFR1信号通路的几种蛋白质(包括 RIPK1、RIPK3、Casp8 和 MLKL)的位点特异性泛素化事件丰度的TSZ依赖性增加(图 4a)。对内源性MLKL的K-ε-GG Ub残余肽的检查揭示了K51、K77、K172 和K219的TSZ依赖性泛素化(图 4b、c)。此外,泛素化残基被定位到4HBD(K51和K77)、第二个brace螺旋(K172)和假激酶结构域(K219)。值得一提的是K219,该残基在静止条件下与激活环的Q343形成氢键(图 4c),此结果可导致MLKL 8的“激活”。因此,在S345磷酸化后,K219的ε-氨基可用于泛素化。这解释了为什么只有磷酸化的活性MLKL会发生泛素化,同时也增加了K219的泛素化可能稳定MLKL的激活形式的可能性,这有助于MLKL的坏死性凋亡潜能。
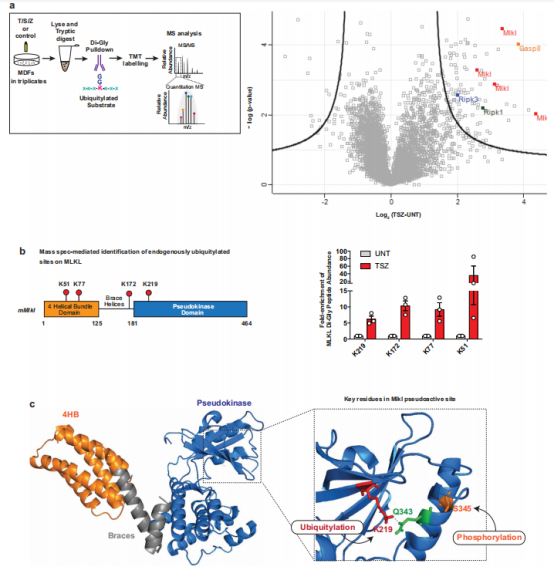
图4
5.K219的泛素化有助于MLKL的细胞毒性潜力
为检测MLKL泛素化的重要性,使用不同的Ub受体赖氨酸突变体重构了Mlkl−/−MDFs。由于Arg具有与Lys相似的化学结构,预计K219R在静止条件下会保持与Q343的氢键(图 5a)。因此,MLKL K219R在静息条件下的表现与 MLKL WT相当,并且它们的表达在没有坏死性凋亡刺激的情况下具有良好的耐受性和非细胞毒性(图 5b)。在用TSZ处理后,MLKL K219R在诱导坏死性凋亡方面的效力明显低于WT MLKL(图 5c),表明了K219的泛素化有助于MLKL的细胞毒性潜力。与K219相比,突变位于MLKL 4HBD的Ub受体赖氨酸K51和K77(MLKL K51R、K77R)对MLKL的杀伤潜力没有影响,无论是单独作用还是联合作用(图 5c)。进一步研究发现MLKLS345D、K219R诱导坏死的作用明显低于MLKLS345D(图5d),这一结果再次证明了K219的泛素化有助于 MLKL的促坏死性凋亡潜力。
为解释K219泛素化增强细胞死亡的机制,对未修饰的MLKL (WT)、在S345磷酸化的MLKL (S345phos)、在S345磷酸化和在K219单泛素化的MLKL进行了从mMLKL (PBD: 4BTF) 8的晶体结构开始的全原子分子动力学(MD)模拟(K219ub S345phos)(图5e-i)。总体MD模拟表明S345磷酸化后激活环螺旋和4HB结构域的构象变化,导致MLKL激活。此外,Ub与K219的结合似乎会影响P-MLKL的4HB结构域灵活性,这可能是MLKL聚合的一个重要特征。
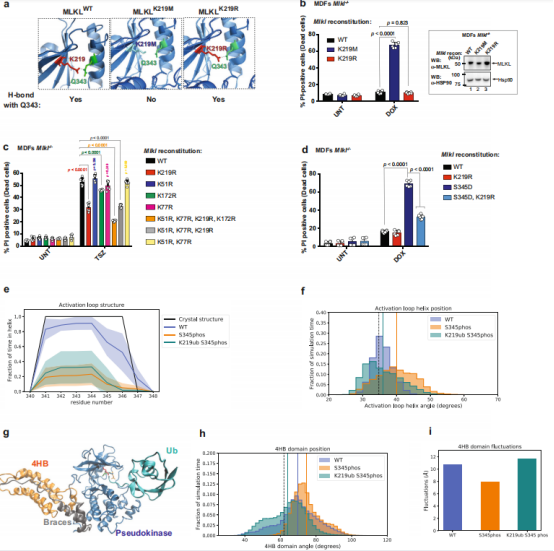
图5 (a) PYMOL模型描绘MLKL突变体氢键的形成; (b) 碘化丙啶阳性 (PI + ) Mlkl -/-小鼠真皮成纤维细胞(MDF)的定量; (c) Mlkl -/- MDF用指定的MLKL泛素突变体进行重组; (d) PI + Mlkl-/- MDFs的定量用指定的MLKL突变体重建; (e) 分子动力学 (MD) 模拟中激活环残基的结构,阴影区域代表从独立模拟计算的标准偏差,线代表平均值; (f) 模拟中激活环螺旋线和相邻螺旋线之间角度的直方图,黑色垂直虚线代表晶体结构中的角度,垂直实线代表每个构建体的 MD模拟的平均角度(WT和K219ub P-S345phos模拟的线重叠); (g) MD模拟的泛素化和磷酸化MLKL,假激酶结构域(蓝色)、brace螺旋(灰色)、4HB结构域(橙色)、Ub部分(蓝绿色)和残基K219(红色)、Q343(绿色)和S345(橙色); (h) MD模拟中4HB结构域与假激酶结构域之间的夹角直方图,黑色垂直虚线代表晶体结构中的角度,垂直实线代表每个构造的MD模拟的平均角度; (i) 4HB结构域相对于假激酶结构域的总体波动。
6.Mlkl K219R/K219R细胞免受TNF和MCMV驱动的坏死性凋亡
从内源性Mlkl基因组位点生成了表达MlklK219R的基因敲入小鼠以研究MLKL在K219位点的泛素化在坏死、病毒感染和组织损伤调控中的作用。首先,通过测量TSZ诱导的骨髓源性巨噬细胞(BMDMs)和MDFs(分别从两对不同的MlklWT/WT和Mlkl K219R/K219R小鼠中分离)的细胞死亡以研究基因敲入突变对坏死性凋亡的影响,结果表明,在K219处消除MLKL泛素化可以防止MLKL介导的细胞死亡(图6a-c)。通过评估K219的泛素化是否有助于在坏死性凋亡过程中在质膜上形成高阶MLKL聚合物,发现虽然用TSZ处理刺激了WT细胞膜上大的MLKL聚合物的形成(图6d),但即使MLKL K219R/K219R被磷酸化这种聚合物也几乎没有在Mlkl K219R/K219R MDFs中形成(图6c, d)。此外,通过用MCMV WT和 MCMVM45mutRHIM感染细胞来探索K219泛素化在MCMV诱导的细胞死亡中的作用,结果显示,用MCMVM45mutRHIM感染很容易诱导WT MDFs细胞死亡(图 6e),相比之下,Mlkl K219R/K219R MDFs 不受MCMV诱导的坏死性凋亡的影响(图 6e)。在研究K219泛素化对病毒生长的影响中,发现Mlkl K219R/K219R MDFs 在感染后72小时后具有比WT 细胞显著更高的病毒滴度(图 6f),实验证实了K219突变本身不影响病毒感染(图 6f)。
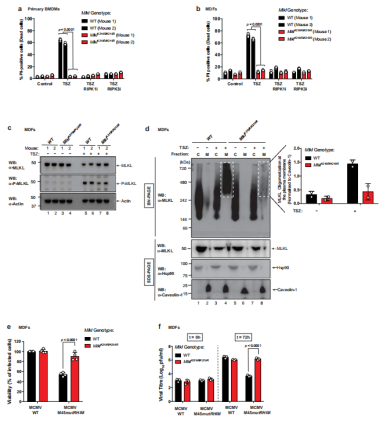
图6
7.Mlkl K219R/K219R基因敲入小鼠免受坏死性凋亡诱导的组织损伤
评估WT和Mlkl K219R/K219R敲入动物皮肤中坏死性凋亡的影响,发现皮下注射临床IAP拮抗剂ASTX660联合泛半胱天冬酶抑制剂Emricasan (AE处理)导致MlklWT/WT动物皮肤严重溃疡(图7a–c),相比之下,Mlkl K219R/K219R敲入动物明显免受AE诱导的组织损伤,这与对照Mlkl -/-动物相当(图 7a-i)。在WT动物中皮下注射AE导致表皮溃疡,在下面的真皮中形成致密的胶原纤维组织,似乎延伸到皮下组织(图 7b-i)。此外,Mlkl K219R/K219R和Mlkl -/-动物表现出没有溃疡的表皮层(图 7b),在Mlkl K219R/K219R和Mlkl -/-小鼠中也没有坏死的证据(图 7b),虽然Mlkl K219R/K219R显著免受皮下注射AE的影响,但这些动物确实显示出一些不规则的表皮(图 7d),原因是表皮增厚(图 7e)。然而,与WT动物相比,AE的皮下注射仅对Mlkl K219R/K219R动物产生相对较小的影响(图 7i)。这些数据表明皮下注射AE诱导RIPK1驱动的坏死性细胞死亡表型,该表型在Mlkl K219R/K219R小鼠中丢失,表明Ub与K219的结合有助于皮肤坏死性凋亡介导的组织损伤。
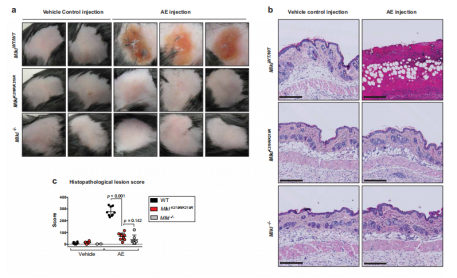
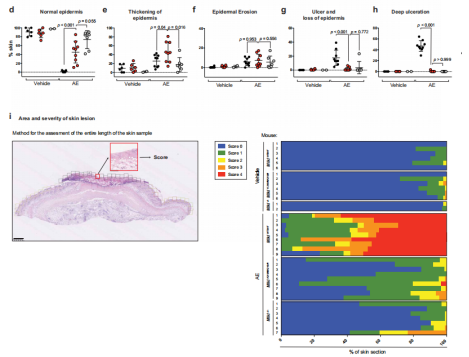
图7
结论
K63连接的泛素链在坏死性凋亡期间连接到MLKL,并且MLKL在K219处的泛素化显著促进了磷酸化MLKL的细胞毒性潜力。K219R MLKL突变保护动物免受坏死性凋亡诱导的皮肤损伤,并使细胞对病原体诱导的坏死性凋亡具有抗性。该项研究证明了MLKL在K219处的泛素化是MLKL在膜上的高阶组装所必需的,从而促进其破裂和坏死,此外,K219泛素化许可 MLKL活性以诱导裂解细胞死亡,这表明病原体的坏死性清除以及MLKL依赖性病理受泛素信号系统的影响。

